





Abstract
The term ‘ancient DNA’ (aDNA) is coming of age, with over 1,200 hits in the PubMed database, beginning in the early 1980s with the studies of ‘molecular paleontology’. Rooted in cloning and limited sequencing of DNA from ancient remains during the pre-PCR era, the field has made incredible progress since the introduction of PCR and next-generation sequencing. Over the last decade, aDNA analysis ushered in a new era in genomics and became the method of choice for reconstructing the history of organisms, their biogeography, and migration routes, with applications in evolutionary biology, population genetics, archaeogenetics, paleo-epidemiology, and many other areas. This change was brought by development of new strategies for coping with the challenges in studying aDNA due to damage and fragmentation, scarce samples, significant historical gaps, and limited applicability of population genetics methods. In this review, we describe the state-of-the-art achievements in aDNA studies, with particular focus on human evolution and demographic history. We present the current experimental and theoretical procedures for handling and analysing highly degraded aDNA. We also review the challenges in the rapidly growing field of ancient epigenomics. Advancement of aDNA tools and methods signifies a new era in population genetics and evolutionary medicine research.
1. Ancient DNA as an indispensable source of information
The passion for unravelling and reconstructing the history of life on Earth has always stimulated research in evolutionary biology. Although inferences of past events such as the states of ancestral organisms (e.g. ancestral sequences), evolutionary episodes (e.g. speciation), and the dynamics governing change (e.g. mutation models) can be obtained through computational phylogenetic and coalescent approaches using contemporary data, naturalists have always valued direct observation above all other methods. Ancient DNA (aDNA) is thus expected to revolutionize evolutionary genetics in the same manner that systematic approach to the analysis of fossil records revolutionized palaeontology: it is a direct window into the past — a ‘time capsule’. aDNA has already been invaluable in addressing many key questions in evolutionary biology, 1–14 frequently providing the only available evidence.
Studies over the past several decades have demonstrated that aDNA can survive and be extracted from ancient and historical material (e.g. bones, teeth, eggshells; mummified, frozen, or artificially preserved tissues). The first attempts to extract and analyse aDNA were performed before the PCR era. In a pioneer study in 1984, Higuchi et al. 15 managed to recover DNA using bacterial cloning from dried muscle of quagga, an extinct subspecies of plains zebra ( Equus quagga ). However, due to extremely poor DNA preservation, analyses of aDNA were limited until an effective technology for DNA amplification, like PCR, made very small amounts of DNA accessible for study. In addition, next-generation sequencing (NGS) technologies and the plummeting cost of DNA sequencing have provided an unprecedented opportunity to perform millions of sequencing reactions in parallel. These advances enabled the first report on ancient sequences retrieved by NGS in 2004. 16 Major milestones of development of high-resolution ancient human genomics 1,2,5,11,12,15,17–29 are shown in Fig. 1 .
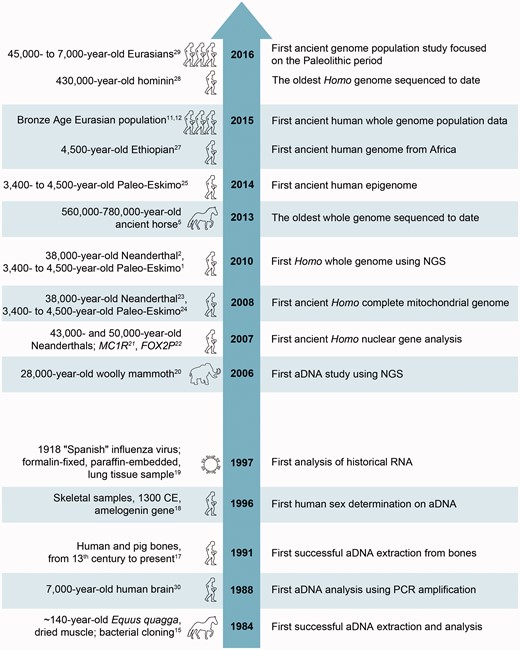
Major milestones of development of high-resolution ancient human genomics.
1.1. Human evolution and demographic history
Over the past decade, genomic techniques have been reshaping our fundamental understanding of human prehistory and origins. 30,31 Until recently, much of what was known about prehistory came from the study of archaeological sites and anthropological investigations, piecing together patterns of human migration and admixture from physical features, pottery, weapons, ornaments, art production, traditional customs, and studies of modern DNA. 32 Other sources of information included linguistic classifications and ancient texts. Although undeniably powerful, these approaches often yielded more questions than answers, and their resolution required incorporation of additional data. Analysis of ancient human remains can reveal migration patterns, 4,10–13 address questions of kinship and family structure, 33 and provide insight into physiological or morphological characteristics such as blood group, skin colour, hair type, 34–37 and climatic adaptation. 2 When combined with other evidence, sequencing ancient genomes could help settle important debates within archaeology or linguistics. This approach, although not infallible, is particularly valuable now when ancient genetics is considered to be a highly robust tool and has significantly impacted many fields such as forensics and history. 38
Sequencing of the genomes from archaic hominids have illuminated earlier events in human evolution and suggested that early hominids had a richer evolutionary history than was previously appreciated. Analysis of Neanderthal genomes extracted from the remains found in Europe and Western and Central Asia and dated 230–30 thousand years ago (kya) demonstrated that contrary to previous suggestions, Neanderthals and anatomically modern humans (AMH) may have interbred. 1,8,39–46 The studies showed that Neanderthals share more genetic material with modern humans across Eurasia than those from sub-Saharan Africa, indicating that genetic flow from Neanderthals to Eurasian AMH likely occurred after the emergence of humans from Africa but before the divergence of Eurasian groups. 1,8 Additional gene flow events may have occurred later in Europe 44 and East Asia. 41,47 Mitochondrial DNA (mtDNA) sequences of morphologically ambiguous Neanderthal bones from Teshik-Tash cave in Uzbekistan and Okladnikov cave in Southern Siberia provided evidence that Neanderthals had an extensive range prior to their extinction. 48 Since the first description of the Neanderthal genome, a number of studies have suggested that various Neanderthal alleles have been preferentially retained in modern populations due to specific selective pressures. 49 Remarkably, the proportion of Neanderthal ancestry in Eurasians decreased substantially since the Palaeolithic, from 4–6% to 1–2% today, suggesting that negative selection against Neanderthal alleles is at work. 28
Genetic analysis of mtDNA from a phalanx dated 48–30 kya recovered from the Denisova cave in Southern Siberia revealed another hominid, named Denisovan, which is genetically distinct from Neanderthals and modern humans. 50 Since then, only two more samples (molars) of Denisovans have been discovered 3 and recently sequenced. 51 Comparison of Neanderthal 1,8 and Denisovan 3,51 genomes suggested that for a long time their population histories were independent of each other. The Denisovan mtDNA represents a deep branch, with the Neanderthal mtDNA closer to that of modern humans 3 . Comparative analysis of the Denisovan and modern human genomes revealed that the genetic contribution from Denisovans to modern humans may have been restricted to Melanesia and Australia with hybridization events taking place mostly in the Southeast Asian mainland, although they may have permeated to Oceania 3,4,50,52,53 as recently suggested by the existence of a widespread, low-level signal of Denisovan ancestry across South and East Asian and Native American populations. 54 However, the exact scenario is hard to identify. 55,56
NGS analysis of a nearly complete mitochondrial genome of a hominid found in Sima de los Huesos cave in Atapuerca, Spain and dated to >300 kya 57 suggested the existence of another branch in the human evolutionary tree. Surprisingly, the Sima de los Huesos mtDNA forms a clade with the mitochondrial genome of Denisovans rather than that of Neanderthals, demonstrating an unexpected link between Denisovans and Middle Pleistocene European hominids. Recently, approximately three million bases of nuclear sequences were obtained from a Sima de los Huesos femur fragment, an incisor, and a molar. 27 In contrast to the mtDNA, the nuclear genomic sequences of Sima de los Huesos are significantly more similar to Neanderthals than to Denisovans. 27 These results agree with previous morphological analyses 58,59 but present an archaeological puzzle.
Studies of aDNA have also delineated human migration routes around the world, particularly in Europe. Analysis of human genomes from Europe and Siberia dated 24–5 kya 7,10,14,34,35 revealed at least three different sources of the population diversity of modern Europeans, i.e. West European hunter-gatherers, ancient North Eurasians with high similarity to Upper Palaeolithic Siberians, and early European farmers originating in the Near East. 7,60
Further aDNA studies allowed mapping migration in Europe in greater detail. A recent study of 69 European individuals who lived 8–3 kya 12 demonstrated that between 8 and 5 kya populations of Western and Eastern Europe were genetically distinct. Groups of early farmers of Near Eastern origin 60 arrived in Western Europe and mixed with local hunter-gatherers, whereas Eastern Europe at that time was inhabited by a distant branch of ancient North Eurasian hunter-gatherers. 10 However, Eastern Europe did not remain a ‘hunter-gatherer’s refuge’ for too long. Around 6–5 kya, farming populations of West Anatolian ancestry appeared in Eastern Europe and mixed with local hunter-gatherers in the Pontic-Caspian region, giving rise to pastoralist people of the extremely successful Yamnaya archaeological culture. Such multiethnic melting pots were fertile ground for many innovations, such as horse domestication and wheeled vehicles from the Yamnaya culture, 61 which probably enabled massive migration or invasions into Western and Northern Europe ∼4.5 kya, introducing their ancestry, languages, and customs. Haak et al. 12 reported that this steppe ancestry persisted in central Europeans from at least 3 kya, and it is ubiquitous in present-day Europeans. At approximately the same time, similar migrations spread Yamnaya-related cultures into South Siberia and Central Asia, as revealed by another large-scale study of 101 genomes from Eurasian Bronze Age (5–3 kya) burial sites. 11 Such large-scale aDNA studies 11,12,60 have not only made technical breakthroughs but also had significant interdisciplinary effects: they influenced the decades-long debate in archaeology and linguistics about the origin of Indo-European language speakers and shed light on perennial questions about the prevalence of traits like skin colour and lactose intolerance in modern Europeans.
With the progress in aDNA sequencing technology, notable studies of remains from all over the world have begun to emerge, 62–64 shedding light onto, e.g. settlement of China and the Pacific islands. Recently, Liu et al. 65 reported the discovery of an 80,000-yr-old man (the earliest modern human in southern China) raising questions about canonical paradigms of human dissemination, since there is no evidence that humans entered Europe before 45 kya. Sequencing of this individual would provide additional insights into the dispersal of humans in Eurasia. A recent study of a 4,500-yr-old Ethiopian skeleton preserved in relatively cool mountainous conditions was the first example of successful aDNA analysis in Africa, 26 giving hope to forthcoming studies of this incredibly interesting region.
Pending future advances in functional genomics, aDNA might prove an unrivalled source of information on the evolution of traits associated with cognitive phenotypes. For example, discovering the genetic variants responsible for language acquisition may allow researchers to pinpoint the origin of complex language in the human lineage, indubitably a cornerstone event in human evolution. Approximately a decade ago the Neanderthals were found to bear a modern human version of FOXP222 gene (likely responsible for the ability to speak 66 ). The authors suggested that the modern variant of FOXP2 was present in the common ancestor of Neanderthals and modern humans. 22
We can also expect aDNA genomic studies to provide direct evidence about human adaptation substantiating the genetic basis of selection. For example, a genome-wide scan of 230 West Eurasians who lived 6.5–1 kya and their comparison with modern human genomes identified significant signatures of selection in a range of loci related to diet (lactase persistence, fatty acid metabolism, vitamin D levels, and some diet-associated diseases), pathogen resistance, and externally visible phenotypes (skin and eye pigmentation, tooth morphology, hair thickness, and body height). 60 This work demonstrated the utility of aDNA data in human adaptive evolution studies. The currently available set of published human aDNA NGS data, including sample IDs, dating, archaeological cultures, site names and locations, references, and links to data repositories, is in Supplementary Table S2 and illustrated in Fig. 2 .
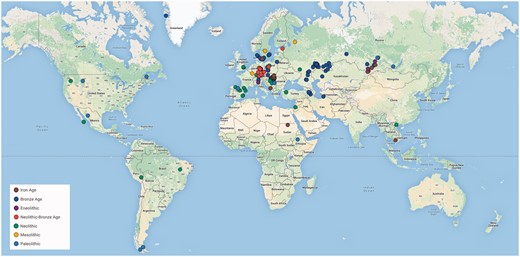
Geographic distribution of existing whole genome aDNA sequences.
1.2. Historic patterns in the spread of infectious diseases
Some devastating pandemics, like the Black Death, remain infamous even centuries after these catastrophes. aDNA enables discovery of the origin and spread of disease-carrying alleles to aid modern epidemiology. Such analyses are possible when genotypes of ancient humans are recovered along with the genomes of their pathogens. For example, Rasmussen et al. 67 sequenced DNA extracted from ancient human teeth and found that Yersinia pestis , the etiological agent of plague, infected humans in Bronze Age Eurasia as early as 5 kya, three millennia before the first historical records of plague. The authors concluded that the bacterium became the highly virulent, flea-borne bubonic plague strain only ∼3 kya by acquiring specific genetic changes. 67
Analysis of Mycobacterium tuberculosis genomes from remains of ancient humans and animals helped in deciphering the origin and dispersal of tuberculosis in human populations. aDNA studies provided support for the hypothesis that the appearance of tuberculosis in humans was not connected to animal domestication as it was suggested before. On the contrary, M. tuberculosis strain in humans is the most ancient one and other tuberculosis strains causing animal diseases evolved from the human strain. 68 Tuberculosis spread with humans and evolved in local conditions. 69,70 The most ancient, so far, human M. tuberculosis strain was discovered in a 9,000-yr-old pre-pottery Neolithic settlement in the Eastern Mediterranean 66 where, in spite of the presence of quantities of bovine bones, no signs of the bovine strain, M. bovis , were found. Discovery of M. bovis strain in human remains from the Iron Age (as well as animal-like Mycobacterium strains in pre-Columbian humans) showed that back-infection from animals took place 6,67 at a later time.
Studying dental plaque of Europeans from different periods (Mesolithic, Neolithic, Bronze Age, Early Medieval, Late Medieval, and present time) demonstrated important shifts in human oral microbiota during recent evolution. The first shift took place in the early Neolithic period with the introduction of farming when more caries- and periodontal disease-associated bacterial taxa were detected. The oral microbiota composition remained stable between the Neolithic period and modern times. Recently, possibly during the Industrial Revolution in the nineteenth century, cariogenic bacteria became dominant, likely due to consumption of industrially processed flour and sugar. Consequently, the genetic diversity of the oral microbiotic ecosystem was impinged, which contributed to the spread of chronic oral and other diseases in countries with post-industrial lifestyles. 71
One of the most remarkable achievements in the field is the study of historical RNA. In 1997, Taubenberger et al. 19 extracted and analysed RNA from the virus that caused the ‘Spanish flu’ pandemic that killed at least 20 million people in 1918–1919. 19,72–76 Reconstruction of the viral genome helped to reveal its origin and discover the mechanism of its exceptional virulence. In contrast to modern influenza viruses, which require an exogenous protease for their replication, the 1918 pandemic virus could replicate without exogenous trypsin. The ‘Spanish flu’ viral genome contained a constellation of genes essential for optimal virulence, which contributed to the strain’s ultra-high virulence. 75 This knowledge enabled epidemiologists to develop a vaccination strategy against another potential pandemic virus. 74
2. Adaptation of experimental and computational methods to the specific biochemistry of aDNA
After the death of an organism, all of its biomolecules are degraded either by host enzymes released from their proper compartments or by saprobic microorganisms. Therefore, compared with modern DNA, aDNA has lower concentration; it is fragmented, contaminated, and chemically modified. 16,77,78 aDNA is also commonly damaged by strand breaks and cross-linking in addition to oxidative and hydrolytic degradation of bases or sugar residues. Relative preservation of DNA in old samples depends on environmental circumstances, such as temperature, humidity, pH, or oxygen, rather than the absolute age of the sample. For instance, DNA samples extracted from frozen remains dated thousands or even hundreds of thousands of years can be of better quality than much more recent samples. 5,79–81 Recent studies showed that the age of ‘readable’ (by current methods) aDNA products is restricted to ∼1–1.5 million years. 11,75 At present, the 560–780 thousand years old Middle Pleistocene horse is the most ancient organism from which reliable aDNA data have been procured. 5 Below, we describe methods for overcoming difficulties caused by each one of the special aDNA features.
2.1. Degradation
Early success with aDNA extraction and sequencing raised hopes that museum specimens, ancient samples, and archaeological finds would provide a plethora of aDNA, but such hopes faded when it became clear that these old samples did not yield any usable DNA. 79 Unfortunately, it is not uncommon for aDNA projects to be disbanded due to low or undetectable DNA content 82–84 . In many other projects, the aDNA concentration is so low that it demands destructive sampling to yield adequate sequencing coverage. That, in turn, results in low genomic coverage (percentage of the length of the reference genome that is covered by mapped reads from the sample) and less reliable genotype calls. In their analysis of Neanderthal DNA, Green et al. reported GC content to be positively correlated (r = 0.49) with retrieval success of sequence fragments, 1,23,85,86 likely due to the faster denaturation of AT-rich regions. They also found G and T overrepresented at the 5′ and 3′ ends of break points and suggested de-purination as a significant cause of strand breaks. 23
Some of the difficulties in working with aDNA were resolved by technological breakthroughs. Improvement of extraction protocols can substantially increase the quantity and quality of aDNA. Thus, modern protocols 4,87 enable extraction and analysis of very short fragments (<50–60 bp, which constitutes the vast majority of aDNA). DNA fragmentation posed difficulties for conventional PCR, which requires amplification of a large number of overlapping fragments to cover a relatively long fragment of DNA, and it is impossible to sequence very short fragments (50–70 bp) using Sanger sequencing. However, NGS technologies generate short reads for any DNA. The average retrieved sequence length in most aDNA projects is 50–100 bp, which is the same order of magnitude as the length of reads produced by many current NGS instruments.
Fragmentation and decay of DNA is a natural occurrence not only postmortem but also in vivo . Spontaneous DNA degradation caused by damaging and mutagenic factors is prevented by DNA repair mechanisms that are not present after death. However, controlled DNA degradation in living organism is implemented during programmed cell death (apoptosis) and differentiation of certain cell types (i.e. erythroid, lens and hair cortical cells). A large family of DNase enzymes performs the DNA degradation vital for proper development and functioning of living tissues. Apoptotic processes leading to these changes and DNA degeneration explain the average length of DNA fragments of 140–160 bp and under extracted from ancient mammoth hairs. 88,89 Many processes leading to DNA degradation, including those that accompany cell and tissue senescence (telomere shortening, error accumulation during DNA synthesis), occur naturally in vivo . Apoptosis finds its continuation in postmortem tissues, leading to further fragmentation of DNA even in favourable conditions for specimen preservation. The detailed biochemistry of processes occurring after death still requires further evaluation, and elucidation of their contribution to aDNA quality might be a promising area for research.
2.2. Contamination
Even after successful DNA extraction, results must always be checked for authenticity. aDNA is often contaminated with some level of exogenous DNA (e.g. DNA from ancient or modern saprotrophic bacteria or fungi), postmortem juxtaposition of organisms, or modern human DNA from the researchers themselves. Naturally, low amounts of aDNA (or its complete absence) in the sample might facilitate the domination of PCR products by exogenous DNA, resulting in the recovery of irrelevant sequences. Indeed, in 1990s, a large number of papers were published reporting DNA sequences from extremely ancient remains such as Miocene plant fossils, 90,91 amber-entombed organisms, 92,93 250-million-yr-old bacteria in salt crystal, 94 and dinosaur bones and eggs. 95–98 In one such case, researchers reported successful extraction and amplification of mtDNA cytochrome b fragment from a Cretaceous Period dinosaur. 95 The sequences differed from all modern cytochrome b sequences. This led the authors to believe that they had sequenced authentic DNA from 80-million-yr-old bones. It was later discovered that those mtDNA sequences were not close to avian and reptilian mtDNAs, as would be expected from their phylogenetic history, but rather to mammalian (including human) mtDNAs. It was thereby suggested that the alleged ‘dinosaur’ DNA was contaminated, presumably by modern human DNA. 99–102 A similar course of events occurred in the study of ancient bacterial DNA supposedly preserved in 250-million-yr-old salt crystals, which turned out to be modern bacterial DNA. 103 In addition to these examples, several other aDNA projects have been impeded by contamination of ancient samples. 98,103–106
To prevent contamination, the experiment must be properly managed, including special requirements for sample collection, sterilization of the working area, DNA authentication, and independent reproducibility. 97,107 These protocols are constantly being refined and improved. For example, in addition to mechanical removal of the upper layer and UV and/or bleach treatment of the sample, a brief pre-digestion step was recently suggested, 108 consisting of short-term sample incubation in an extraction buffer and its subsequent removal. According to the authors, this step alone increases the fraction of endogenous DNA several fold. In general, sequencing preparation step plays an important role in minimizing contamination. When there is sufficient material, sequencing library may be prepared entirely without using PCR, greatly minimizing potential for sample contamination. 109 Recent work actually takes advantage of postmortem modifications to enrich for endogenous versus contaminated sequences. 110
In shotgun sequencing of vertebrate samples, a substantial fraction of the reads comes from contamination with environmental DNA from bacteria and fungi. 2,20,111 Microbial sequences are often remarkably different from target species sequences and thus should be easily flagged by a standard BLAST search against the NCBI non-redundant nucleotide database. This strategy, however, fails to discover most of the microbial sequences that have yet to be sequenced. Therefore, it is not surprising that a large fraction of reads in many aDNA libraries is labelled as ‘unknown’ or ‘unclassified’, mainly due to the unidentified microbial content. 86 Frequently, mapping the shotgun sequencing reads onto the reference genome of the target species (or the closest genome at hand) and discarding all reads below a certain level of similarity is preferred 112 alongside choosing tissues with less microbial DNA. For instance, it has been suggested that hair shafts or avian eggshells contain less microbial DNA than bone, 88,113 but these tissues are not available for most ancient samples. Alternatively, recovery of bacterial or fungal sequences is not very likely for PCR-based capture methods, as primers are designed based on known sequences from the sample’s own species or its close relatives.
The intricacy and method of detecting and removing modern human contamination depends on the distance of the target species from humans. Expectedly, it is much easier to handle distantly related species such as mammoths, penguins, or cave bears than archaic hominids, like the Denisovans and Neanderthals, and particularly ancient modern humans. Moreover, the archaeological material in Europe is usually excavated and later handled, extracted, and sequenced by Europeans, sometimes from the same region. The same is generally true for other territories around the world. When a limited number of loci are sequenced from PCR or cloning products, it is possible to examine alignments visually and to inspect individual polymorphic positions to determine which differences are genuine and which are likely artefacts or contamination; 114,115 however, with reads from shotgun sequencing technologies, automated methods are typically required.
Analysing sequence reads in a phylogenetic framework along with sequences from ancient and extant relatives and outgroups is one of the initial steps to ensure that ancient sequences fit within the acceptable phylogeny and flag probable contamination. For instance, sequences from the mammoth were compared with those of the elephant, its closest kin, and to outgroups, such as humans and dogs, to ascertain phylogenetic correctness. 20 Filtering reads that were mapped onto the elephant genome with a high score and matched the elephant genome better than that of human, dog, or other species helped to remove human and microbial contamination. Neanderthal samples were phylogenetically examined to see if they fall outside the range of modern human variation. 48
Initially, a number of human and non-human studies filtered out samples with long sequence fragments considered evidence of contamination since authentic aDNA is supposed to be fragmented. 82,115 However, it has become clear that the average aDNA fragment length can vary substantially between samples and can overlap with contaminant fragment lengths; therefore, more elegant approaches are needed to develop authentication criteria based on length. In a study of Neanderthal DNA, estimates of human-Neanderthal sequence divergence and the percentage of C→T and G→A (equivalent events) misincorporations did not vary significantly with alignment length. 116 Existence of substantial modern DNA contamination would have produced two types of fragments: authentic ancient ones which were short and had high numbers of mismatches (showed high divergence versus modern human reference), and modern contaminant ones which were long and showed few mismatches (showed low divergence versus modern human reference). Noonan et al. 116 remarked that the absence of an inverse relationship between alignment length and divergence from the human reference meant that the level of contamination with modern DNA was negligible in their data set; however, they did not provide a quantitative estimate. The problem with this approach is that even among authentic ancient fragments, short fragments presumably represent higher rates of base modification and consequently may produce upward-biased divergence estimates. 85
Through the accumulation of ancient sequences over time, positions at which the target sequence (e.g. Neanderthal or Denisovan) have been invariably different from the likely contaminant (e.g. modern humans) can be used to estimate modern DNA contamination. 23,117,118 Here, mtDNA is the marker of choice because of its high copy number, leading to greater sequencing depth; however, the validity of extrapolating mtDNA contamination estimates to nuclear sequences has been questioned based on possible differences in the conservation properties of mtDNA and nuclear DNA. 85 As base modification and misincorporations in aDNA often involve C to U (T) and A to G transitions, contamination with external DNA can be more reliably estimated using transversion or indel counts. 23 Even when sufficient prior data on sequence variation in the archaic hominid population is available, the fraction of reads that deviate from consensus base calls at haploid loci, e.g. those on mtDNA or the Y-chromosome, can provide an estimate of exogenous DNA—assuming that authentic aDNA is more abundant than contamination and that correct sequence reads are more likely than errors. This method is especially applicable to positions at which the modern human population is fixed for the derived base while the archaic consensus base is ancestral. 3,53
Ancient modern humans are not expected to carry informative (fixed) substitutions compared with contemporary humans or necessarily to fall outside the range of modern human phylogeny, although they might do so. A first step in the QC of sequences from ancient modern human samples is to ascertain that all sequence reads come from a single individual. This can be done by estimating X-linked heterozygosity in male samples, Y-linked heterozygosity in male samples, Y-linked presence in female samples, or mtDNA heterozygosity for either gender. 2,53,85,119 Next, it is necessary to show that each specimen in the data set carries unique sequences (e.g. mtDNA or Y-chromosome haplotypes) that are different from sequences of other specimens and from the researchers. 83,120,121
As most of the ancient sequences during the pre-NGS era or shortly thereafter were limited to mitochondrial markers, 23,122,123 it was crucial to distinguish them from nuclear inserts of mtDNA (NUMTs). Generally, a higher alignment score to the mitochondrial sequence than to the nuclear sequences is the authentication criterion. For extinct species without a reference, where sequence reads must be mapped to the genome of another species, this becomes more complicated because the divergence of orthologous sequences must be considered in addition to differences between NUMTs and their mitochondrial counterparts. 23 Considering the low likelihood of heteroplasmy, observing more than one allele with non-negligible frequencies at each position would indicate either external contamination or sequencing of NUMTs.
2.3. Postmortem base modification
Postmortem DNA modifications through hydrolysis and oxidation pose another substantial difficulty for studying aDNA. The most significant alteration is nucleotide deamination, which leads to false transitions during PCR: cytosine to uracil, 5-methyl-cytosine to thymine (both causing incorporation of T instead of C), and, more rarely, adenine to hypoxanthine (causing incorporation of G instead of A). 82,124–127 Chemical modification of nucleotides can lead to reduced sequencing coverage because they prevent mapping of many authentic reads due to an overestimated number of mismatches compared with the reference. They can also result in the erroneous base and genotype calls and false estimates of genomic parameters such as heterozygosity, nucleotide diversity, GC content, or divergence times. Base modifications are often observed in the five to seven final bases of DNA fragments and are thought to occur more readily in the terminal, single-stranded overhangs. 128 These terminal misincorporations are even more problematic because local sequence alignment methods used for mapping the NGS reads onto the reference genome rely heavily on matching initial bases to the reference. 86
To overcome problems with chemical modification, several approaches have been developed. Treatment with uracil- N -glycosylase (UNG) removes uracil residues, thus preventing replication of fragments with deaminated cytosine; 124,129 however, the resulting abasic sites prevent replication by DNA polymerases, which excludes all the fragments with uracil from the reaction. This can be crucial for valuable ancient samples already having low DNA concentrations. A simple modification was suggested recently to overcome this problem: 130 follow-up treatment with endonuclease VIII after UNG repairs most of the abasic sites and enables subsequent analysis of these fragments. This procedure, however, does not resolve the problem of false A→G transitions. Using DNA polymerases such as Phusion ( Pfu ), which does not amplify uracil, also avoids false C→T (but not A→G) transitions but excludes all uracil-containing fragments from amplification, which further decreases the DNA template in the reaction. In addition, since these enzymes can work with methylated, deaminated cytosine (i.e. 5-methyluracil, thymine), the problem remains for methylated aDNA. Single primer extension PCR (SP-PCR) enables analysis of separate DNA strands, which makes it possible both to distinguish real mutations from postmortem modifications and to evaluate the level of these modifications. 124–127,131 SP-PCR is performed in two steps: first, PCR with only one primer is carried out to accumulate only one DNA strand, and then the second primer is added to the reaction and PCR continues with a normal protocol. The resulting PCR product derives mainly from one of the DNA strands. Analysis of these products can identify in which DNA strand postmortem modification occurred. This method requires very thorough selection of PCR primers and annealing temperatures, otherwise non-specific annealing or formation of primer dimers is highly possible.
One estimation strategy for base modification compares the percentage of T and A calls at ultra-conserved C and G positions, respectively. These genomic positions are expected to have retained their ancestral state in the ancient sample, so transitions exclusively observed in the ancient sample can be attributed to base misincorporations. 4,89 Another method compares the frequencies of different types of transitions and transversions in ancient–modern and modern–modern sequence alignments of closely related species (e.g. Neanderthal–human, Neanderthal–chimp, and human–chimp). An excess of C→T (and G→A, respectively) transitions in modern–ancient alignments provides an estimate of base modification. 86 The third strategy takes advantage of the direction of transition induced by base modification. In a 2006 study, the C→U modification in mammoth DNA caused the apparent rate of (mammoth T) → (elephant C) transitions to be 1.9-fold larger that of (mammoth C) → (elephant T) transitions. 20 Recently developed experimental protocols, such as pre-treatment of aDNA with UNG, have reduced the magnitude of this problem.
If the level of base modification is non-negligible, steps must be taken to eliminate or lessen its effect on the output of downstream population genetic analyses. Sometimes, C→T/G→A or all transitions are simply left out of the analyses, and only transversions and indels are included in the calculation of divergence or reconstruction of phylogeny. 1 Alternatively, it is possible to polarize polymorphisms into ancestral and derived states using an outgroup (e.g. chimp for human–Neanderthal comparisons) to place the mutation events on the corresponding branches of the phylogenetic tree using a parsimony approach (of which the branch leading to the ancient sample will probably contain disproportionately high numbers) and to calculate divergence times using information from branches leading to modern samples only. 86 Another strategy takes advantage of the observation that most of the base modifications occur at the 5′ and 3′ ends of fragments and trims a few (5–7) bases off either end of each sequence read to lower the chance of including a misincorporated base. 2,119
Although the problems associated with aDNA anomalies are not insurmountable using available experimental and bioinformatics technologies, drastic variations in the type and magnitude of damage among ancient remains make it impossible to develop a universally successful protocol for aDNA extraction and sequencing. For instance, the fraction of authentic Neanderthal mtDNA among six examined ancient samples varied from ∼1% to ∼99%, 86 and the level of contamination in five well-preserved human bone specimens dated 800–1600 CE varied from 0% to 100%. 120 The Neanderthal mitochondrial genome and partial nuclear genome were retrieved using data from several sequencing attempts. 48,86,116 This compendium was crucial in determining design parameters for assembling the full Neanderthal nuclear genome. 8 The contaminating sequences in an ancient maize microsatellite genotyping project were found to be of different natures across samples: some exhibited mainly microbial contamination, whereas others contained copies of transposable elements. 82 Therefore, an initial round of extraction and sequencing is recommended to estimate quality parameters for each sample (e.g. yield, chemical modification, % contamination, % uniquely mapped reads, and % genome covered) to inform appropriate experimental and data preparation strategies. It is also important to remember that both experimental and computational methods of the overcoming of aDNA problems have advantages and limitations. Therefore, to achieve the most reliable results, it is of great importance that researchers use both these approaches to examine aDNA ( Table 1 ). One of the good examples of the combination of novel experimental and computational approaches, as well as of the good correspondence of the methods to the goal is the paper of Haak et al. 12 where they employed powerful experimental protocols, stringent quality control procedure, as well as bioinformatics and population genetics approaches to test hypotheses about the steppe origin of Indo-European languages carriers.
Difficulties of working with ancient DNA and specialized methods developed to address them
| Problem | Experimental solutions | Bioinformatics solutions |
|---|---|---|
| Degradation |
| Algorithms based on genotype likelihoods rather than a single best genotype for low coverage genomic positions |
| Base damage |
|
|
| Contamination |
|
|
| Problem | Experimental solutions | Bioinformatics solutions |
|---|---|---|
| Degradation |
| Algorithms based on genotype likelihoods rather than a single best genotype for low coverage genomic positions |
| Base damage |
|
|
| Contamination |
|
|
The solutions aimed at one or more of the problems are not mutually exclusive and are often used in combination for better results. In addition, various bioinformatics ideas for tackling contamination and base damage are sometimes integrated into a single Maximum Likelihood framework for base and genotype calling.
Difficulties of working with ancient DNA and specialized methods developed to address them
| Problem | Experimental solutions | Bioinformatics solutions |
|---|---|---|
| Degradation |
| Algorithms based on genotype likelihoods rather than a single best genotype for low coverage genomic positions |
| Base damage |
|
|
| Contamination |
|
|
| Problem | Experimental solutions | Bioinformatics solutions |
|---|---|---|
| Degradation |
| Algorithms based on genotype likelihoods rather than a single best genotype for low coverage genomic positions |
| Base damage |
|
|
| Contamination |
|
|
The solutions aimed at one or more of the problems are not mutually exclusive and are often used in combination for better results. In addition, various bioinformatics ideas for tackling contamination and base damage are sometimes integrated into a single Maximum Likelihood framework for base and genotype calling.
3. Analysis of aDNA data
3.1. Software tools for pre-processing of aDNA NGS data
An important consideration for the analysis of aDNA, which typically undergoes many rounds of amplification, is the presence of PCR duplicates, which must be identified, and ideally removed. 132 Once the sources of contamination or base misincorporation are detected and removed from the aDNA sequences, it is possible to infer genotypes for further analysis. In addition to regular NGS data quality control and pre-processing steps, application of specialized tools is required to address the special features of aDNA. Genotypes can be inferred more accurately by combining observed read bases with various estimators of contamination, base modification, sequencing error, and read alignment quality combined via a single maximum likelihood (ML) calculation. 133 The ML model can be designed in the haploid mode for mtDNA, or X- and Y-chromosomes in males, or diploid mode for autosomal markers or X-linked markers in females. 2 Depending on the specific design, ML models can use these estimators to output the genotype or co-estimate all of these parameters simultaneously.
Current NGS analyses of aDNA are performed with well-established but non-specialized computational tools as novel customized tools for aDNA analysis have not yet been widely accepted, and custom scripts have to be written to adjust for aDNA specifics. Base calling is frequently performed with Illumina’s standard base-caller Bustard BayesCall 134 (flexible model-based tool) and freeIbis 135 (utilizing a multiclass Support Vector Machine algorithm). FastQC 136 is typically used for preliminary quality control of reads. AdapterRemoval, 137 CutAdapt, 138 and SeqPrep 139 are currently the most common tools in the aDNA world for de-multiplexing, adapter trimming, low-quality call trimming, and paired-end merging. Burrows-Wheeler Aligner (BWA) and Bowtie are commonly used for mapping of aDNA reads. 140 Since BWA and Bowtie were developed for high-quality modern DNA reads, parameter adjustments to reflect properties of a DNA must be done. For example, it may be advisable to trim likely damaged positions, disable the seed, adjust gap openings and penalties, and permit indels at read ends. 140 Genome Analysis Toolkit (GATK) 141,142 or SAMtools 143 are then used for variant calling. Methods should be optimized for shorter (17–35 nucleotide) reads with possible adapters on both ends of the read and a large overlap between paired reads, and the call corresponding to the highest quality score should be selected at each position. In addition, due to low quantities of endogenous DNA, the high number of short reads, and high levels of contamination, masking repeat regions may improve read mapping. Several pipelines (e.g. aLib 144 and PALEOMIX 145 ) incorporating all these changes were designed for aDNA analysis. Sometimes selected elements of such pipelines are combined to achieve optimal performance. For instance, the leeHom module of aLib is used to pre-process reads 146 while Anfo, 1 MIA, 117 and BWA-PSSM 147 are used for subsequent read mapping.
After read pre-treatment and alignment, the text file containing the sequence alignment data (termed the SAM file or BAM for binary files) can be used to estimate contamination and degradation levels using tools such as mapDamage 148 and mapDamage2.0. 149 PMDtools 150 (identification of those DNA fragments that are unlikely to come from modern sources) and Schmutzi 151 (maximum a posteriori estimator of mitochondrial contamination for ancient samples) are utilized to select reads for re-analysis that have higher chances of coming from aDNA. Depending on the situation, analysis can be done in a fully automated cycle for the entire genome or only for mtDNA. Such algorithms report the probabilities of different types of postmortem DNA degradation, which allows for better statistical modelling at the variant calling stage, employing SNPest 152 or custom scripts. A typical NGS pipeline for aDNA analysis is shown in Fig. 3 .
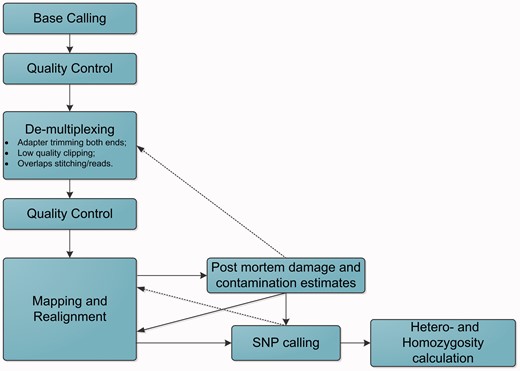
Flowchart of a typical bioinformatics pipeline for aDNA analysis using NGS data.
The amount of extracted endogenous DNA may allow satisfactory coverage of aDNA sequences (as high as 10–20x coverage at a subset of regions for a few samples), comparable to modern DNA studies. Nevertheless, it is very common for ancient samples to have ∼1× average coverage. In such cases, population genetics analysis can still be done using ADMIXTURE and other standard tools by choosing the variant with the highest number of supporting reads (or with the highest quality) instead of trying to make heterozygous/homozygous calls at each autosomal position (which is tricky when there are <5 reads covering a given position resulting in 3–4 conflicting variants). This method can be used when the amount of contaminating modern human DNA is much lower than the amount of endogenous DNA.
Special care needs to be exercised when combining SNP data from ancient and modern samples. Recently published analysis of the first ancient African genome 26 presented an erroneous conclusion that genomes of individuals throughout Africa contain DNA inherited from Eurasian immigrants. 153 The error was noticed, and the authors published an erratum stating that it had been necessary to convert the input produced by SAMtools to be compatible with PLINK, but this step was omitted causing the removal of many positions homozygous to the human reference genome. 153 This example illustrates the importance of using validated pipelines for aDNA analysis.
3.2. Interpretation of aDNA data
Below, we discuss the analytical methods for biologically relevant interpretation of aDNA data. Various population genetics analytical methods have been applied to infer past demographic events based on data obtained from aDNA studies. One of the basic methods for identifying ancient haplotypes is scanning present-day populations for variants identified in the aDNA. This simple approach provides an estimate of populations/regions that harbour such ancient genetic signatures and has been successfully applied to identify modern European populations with mtDNA mutations that were found in aDNA samples. 154,155 Analysis of single-nucleotide polymorphisms (SNPs) in prehistoric samples can shed light on ancestral phenotypes, including pigmentation of skin, hair, and eyes, 37 and the sex of the sample can be computed as the ratio of reads mapping to the Y- and X-chromosomes. 156,157 In the case of uni-parental markers such as mtDNA variants and Y-chromosome markers, the mutational distance between the ancient and modern haplotypes is visualized using phylogenetic network analysis programmes. 155,158 Network analysis of haplotype data reveals genetic distance, mutation rate, and regions of haplotype spread. Recently, a novel method for dating ancient human samples was developed. 159 The method is based on a recombination clock and shared history of Neanderthal gene flow into non-Africans. 159
With increased numbers of recovered ancient and historic DNA samples and steady improvements in aDNA sequencing technology, scientists can study the distribution of ancient human genetic variation and compare it to that of modern populations 160 or gain a deeper level understanding of the distribution of genetic variation within populations by applying admixture-based tools for joint analysis of modern and ancient samples at a population level. Tools and approaches (such as PCA, 161 STRUCTURE, 162 ADMIXTURE, 163 SPAMIX, 164 SPA, 165 ADMIXTOOLS, 166 GPS, 167 LAMP, 168 HAPMIX, 169 reAdmix, 170 MUTLIMIX, 171 mSpectrum, 172 SABER, 173 and others) which were initially developed for population analysis of contemporary individuals, can be applied in combination with anthropological data and historical records to reconstruct migration patterns, provenances, and local and global ancestries of extinct populations. ADMIXTURE is a computational tool for ML estimation of individual ancestries from multi-locus SNP genotype data sets. Recently, Allentoft et al. 11 inferred the ancestral components from modern samples and then projected the ancient samples onto the inferred components using the ancestral allele frequencies inferred by ADMIXTURE. Comparison of admixture profiles of ancient and modern populations within a given region informs the generation of hypotheses about population migrations that can be validated with independent sources and methods of analysis. NGSADMIX uses genotype likelihoods instead of called genotypes to resolve ancestry, which is particularly useful considering the myriad sources of uncertainty in aDNA NGS data. 174 GPS algorithm determines the provenance of an individual—a point on a globe where people with similar genotype live. Tools like SPAMIX and reAdmix model individual as a weighted sum of reference populations.
To infer the geographical origin of a specific haplotype, it is essential to partition the genome into haplotypes with distinct ancestries that may have been inherited from multiple populations. Such haplotypes can be obtained using ‘local ancestry’ tools (HAPMIX, or SABER, LAMP, and MULTIMIX and others) which allow inference of ‘local ancestry’ instead of the ‘global ancestry’ (that can be inferred with PCA, SPAMIX, GPS, ADMIXTURE, STRUCTURE, reAdmix) and their usage depends on the complexity of the data set, the expected mixture levels, and the available phenotypic data. For instance, if the phenotype is associated with a particular trait, a ‘local ancestry’ tool is preferred, whereas a ‘global ancestry’ tool should be used when the phenotype is a complex trait involving multiple unknown loci. The list of the described software tools is shown in Supplementary Table S1 .
When several individuals of an ancient population are available, certain population genetic parameters can be estimated. For example, by examining a number of microsatellite loci in 160- to 200-yr-old Daphnia samples, 175 researchers were able to calculate the heterozygosity, gene diversity, deviation from Hardy–Weinberg equilibrium, and linkage disequilibrium between pairs of markers. An analysis of 21 samples from a graveyard in Germany dated ∼6 kya allowed analysis of the mtDNA and Y-chromosomal haplotype diversity as well as the selection forces inferred from Tajima’s D. 83 Jaenicke-Despres et al. 176 discovered allelic variants of three genes that differentiate modern maize and teosinte from 11 maize cobs dating to 660–4,400 yrs ago, opening a window to the genetic chronology of maize domestication. It should be noted, that quantitative data, such as allele frequencies from multiple poorly preserved sample should be treated with caution, as postmortem DNA degradation can bias allele frequency estimates. 109 However, population genetics also offers a range of neutrality tests, such as Hardy–Weinberg equilibrium, which can be used to check for the presence or artefactual sites, when compared with the present-day data.
Even when only a single member of an ancient population can be recovered, a number of genomic and evolutionary inferences can be made about its taxon or population. For example, based on remarkably low levels of dN/dS (non-synonymous to synonymous substitution ratio), it was concluded that mitochondrial proteins were under strong purifying selection in Denisovans 50 . Conversely, the higher dN/dS ratio calculated from Neanderthal genomes was attributed to smaller effective population size and inefficient purifying selection 23,117 The heterozygosity of the TAS2R38 locus in a single Neanderthal individual was used to infer that he was a bitter-taster and, further, that this trait varied among Neanderthals 177 correlation along the genome, it was suggested that the Denisovans experienced a 30-fold decrease in effective population size compared with African humans. 178 Sporadic calculations from museum data can likewise be extremely useful in inferring population history, e.g. for determining genetic continuity of populations. 179
4. Beyond DNA sequence: ancient epigenomics
Many human phenotypes, including physical and psychological characteristics and predispositions to chronic diseases, arise from complex patterns of gene expression, which are, in turn, influenced by poorly understood interactions of so-called ‘genetic determinants’ and external environmental signals. These intricate interactions are commonly explained by (equally poorly understood) epigenetic mechanisms including DNA methylation, histone modifications, and a spectrum of non-coding RNAs that modify the structure of chromatin and modulate gene expression. Until recently, reconstructing the gene expression profile of specific postmortem samples using only DNA was deemed impossible. Several research groups analysing the methylation maps (methylome) of Neanderthals proposed that patterns of CpG methylation could be preserved in the DNA. 130,180 In 2012, Llamas et al. 181 applied bisulphite allelic sequencing of loci to late Pleistocene Bison priscus remains and demonstrated preservation of methylation patterns, although postmortem deamination of methylated cytosine to thymine prevented accurate quantification of methylated cytosine levels.
In 2014, another approach for genome-wide methylation studies of ancient samples was suggested. 25,180,182 In bisulphite sequencing, unmethylated cytosines are chemically converted into uracils, which are then amplified by some polymerases, such as Taq , as thymines (T), while mC is unaffected and is amplified as C. In postmortem samples C→U and mC→T spontaneous conversions occur naturally. To discriminate between C→U and mC→T, the same fragments must be amplified by two different polymerases ( Fig. 4 ). Taq DNA polymerases can replicate through uracils, while high-fidelity DNA polymerases, like Phusion ( Pfu ), cannot. Thus, it is possible to detect methylated cytosines in aDNA by their elevated C/T mismatch rates as compared with unmethylated cytosines. However, analysis of aDNA methylation is limited to ∼10 nucleotides from the fragment’s ends. There are two reasons for this. First, the probability of deamination drops exponentially with the distance of a C nucleotide from the fragment’s end. 130 Second, the further the methylated C is located from the fragment’s end, the higher the probability that Pfu will encounter (and will fail to bypass) uracil originated from the conversion of unmethylated cytosine and will not reach C.
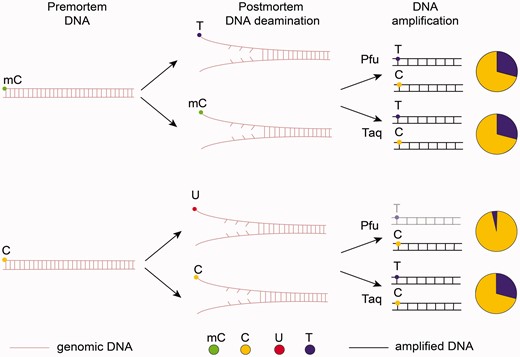
Epigenetic analysis of aDNA. As a result of cytosine and methyl-cytosine deamination in postmortem sample, we observe C→U and mC→T conversions. When Taq polymerase is used for DNA amplification, both C→U and mC→T will be recorded as T (this is the major difference between ancient and bisulphite-treated samples when only unmethylated cytosine in converted to U while mC remains unchanged). When Pfu polymerase is used, U will not be amplified, while those T that appeared as a result of mC→T conversion will be read as T. The pie charts demonstrate the ratio of sequenced C to T. This C/T ratio with Taq and Pfu along with comparison with the reference genome allows detection of methylated cytosines: in the case of postmortem deamination C→U and PCR by Pfu the frequency of T will be decreased.
The described strategy was applied to analyse aDNA from Neanderthal (50 kya), Denisovan (40 kya), and a relatively recent Palaeo-Eskimo individual (4 kya). Overall, DNA methylation patterns in ancient human bones or hairs were almost indistinguishable from those in modern humans. However, by examining differentially methylated regions, Gokhman et al. 180 found that some key regulators of limb development, like HOXD9 and HOXD10 , had methylated promoters (in Neanderthal) and gene bodies (in Denisovan), whereas these regions are hypo-methylated in bones of present-day humans. Deregulation of the HOXD cluster genes results in morphological changes in mice. 183 Since this deregulation corresponds to Neanderthal–modern human differences, it can be inferred that epigenetic changes in the HOXD clusters might have played a key role in the recent evolution of human limbs. Differentially methylated regions were also found within the MEIS1 gene, which encodes a protein that controls the activity of the HOXD cluster. 183
Interpretation of ancient methylome from aggregated C/T mismatch information over large genomic regions allows to determine whether extended regions with altered DNA methylation were present in ancient samples. These include not only hypermethylated CpG islands but also (i) large (from 10 5 to 10 6 bp long) partially methylated gene-poor domains that co-localize with lamina-associated domains; 184,185 (ii) DNA methylation valleys extending over several kb of DNA, which are strongly hypomethylated in most tissues, enriched in transcription factors and developmental genes; 186,187 (iii) undermethylated canyons (up to dozens of kb) that were recently identified in hematopoietic stem cells; 188 and (iv) epigenetic programmes associated with intestinal inflammation and characterized by hypermethylation of DNA methylation valleys with low CpG density and active chromatin marks. 189
Methylation analysis typically focuses on genomic regions that span several kb or even mb. 187–189 The C→T mismatch aggregation strategy, applied in a recent aDNA epigenomic study, 25 could yield new perspectives on adaptation signals and disease markers if soft tissues are found. Brain, intestine, muscle, and blood are not normally preserved in anthropological samples, and extreme conditions (such as permafrost soil) are required for preservation. Analysis of epigenetic patterns also allows estimation of the individual’s age at death using a recent forensic study that found a correlation between the methylation state of specific CpGs and the age of an individual. 190 Such calculations are based on the assumption that environmental signals 6 kya produced the same genomic methylation response observed today to estimate the age of ancient humans using modern databases. Using this approach, Pedersen et al. 25 calculated that the Saqqaq individual was probably in his late thirties when he died.
Methylated CpG’s are almost exclusively found in vertebrate somatic cells; bacterial genomes feature methylated cytosines and adenines but rarely in a CpG context. Hence, CpG methylation levels can be used to enrich the endogenous content of a human aDNA sample and separate it from bacterial contaminants. 191 Methyl DNA binding domain (MBD) affinity chromatography, allowing separation of methylated DNA probes containing a single methylated CpG, has become a routine method for establishing methylomes of genomes of different origins. 192 Application of this method to aDNA can facilitate characterization of ancient methylomes and separate vertebrate and microbial fractions of aDNA extracts. Using the remains of the Saqqaq Palaeo-Eskimo individual, woolly mammoths, polar bears, and two equine species, methylation marks were shown to survive in a variety of tissues and environmental contexts and over a large temporal span (>45–4 kya). Additionally, MBD enrichment allows microbiome characterization for ancient samples and potentially reconstruction of genomes of ancient pathogens.
Although DNA methylation may serve as an indicator of gene silencing, epigenetic analysis alone is insufficient to determine whether the gene was destined for transcription or silencing. Additional data, such as histone modification marks, chromatin structure, and transcription factor binding information, are essential for gene activity prediction. Even though research on ancient proteins is at the nascent stage, shotgun sequencing of aDNA provides a surprisingly rich source of epigenetic information. Pedersen et al. 25 observed unexpected periodicity in the density of covered nucleotides along the Saqqaq genome and hypothesized that these periodic patterns could stem from the protection of DNA by nucleosome binding with preferential degradation of linker regions between nucleosomes. Under this scenario, the observed read depth would reflect the nucleosome occupancy. Analysis of the spectral density (periodogram) in transcription start site (TSS) regions showed that the frequency spectrum has a peak in the relative signal at 193 bp corresponding to the expected inter-nucleosome distance. 25 Moreover, a phasogram from Fourier transform revealed a short-range (10 bp) periodicity, reflecting preferential shifts in nucleosome positioning every 10 bp and/or preferential cleavage of the DNA backbone facing away from nucleosome protection. 193 Strongly positioned nucleosomes in an ancient sample were also found within the vicinity (4 kb) of the transcriptional repressor CCCTC-binding factor (CTCF) binding sites, and their order was negatively correlated with uncovered DNA methylation. 25 Since DNase I-hypersensitive sites (DHSs) near the TSS are reliable predictive markers for gene transcription, 194 regions within open chromatin structures may be more susceptible to postmortem or apoptosis-induced DNase cleavage, in which case the density of NGS reads near the TSS of active genes would be lower than at silent genes. Based on read density at known TSSs and DHSs from the ENCODE project and using de novo methods of TSS prediction (e.g. NPEST 195 or TSSer 196 ), it is possible to sort TSSs according to transcriptional activity of corresponding genes. In the near future, it may be feasible to quantitatively reconstruct gene expression patterns of ancient samples by combining nucleosome positioning, the presence of DHSs at TSSs, and DNA methylation. Therefore, analysis of preserved brains, such as those from bog bodies, 197 will be of particular interest. Recently found remains of a woolly mammoth that retained brain structures of a very high quality 198 raised hopes that exciting discoveries are on the horizon that would allow us to test whether the higher nervous system activity in modern humans differs from that of ancient humans at the epigenetic level. 199,200
Conclusions
aDNA research has revolutionized a multitude of scientific disciplines. Representing the most direct route to address a large number of questions in evolution, medicine, anthropology, and history, aDNA became an indispensable tool in population genetics, paleo-epidemiology, and related fields. Analysis of aDNA has made tremendous progress since its humble beginning in the early 1990s, when contamination with modern DNA sources was commonplace, and only limited analysis was possible due to DNA fragmentation and sparse sampling. In this review, we attempted to provide a detailed overview of recent innovations aimed at coping with these limitations, both through experimental procedures and bioinformatics algorithms. We also considered challenges regarding aDNA biochemistry and degradation, particular bioinformatics tools compensating for short reads and gaps in sequencing coverage, and advances in population genetics to handle sparse sampling. Finally, we described the particularities of aDNA epigenetics and functional interpretation of deduced activities of genes and pathways.
In envisioning future progress in aDNA studies, we would like to note that not every advance in genomics or experimental biology may affect the field. Recent breakthroughs in genomic technologies drastically increased the amount of information obtained from aDNA, and new inventions, e.g. progress in targeted enrichment methods and single-molecule sequencing, would likely allow investigation of previously intractable samples from hot climates and more distant eras. However, experimental approaches will always be limited by the quantity and quality of aDNA in ancient remains. Thus, development of computational methods to cope with aDNA-specific biases and extract meaningful information from low-coverage aDNA data is critical. Studies of aDNA will hugely benefit from further improvement of sophisticated bioinformatics tools coupled with the rapid accumulation of content (reference genomes and variant databases) from both ancient samples and freshly sequenced modern human populations. Regarding the latter, one can hardly overestimate the effect of international projects on systematic genotyping and sequencing of small and/or remote human populations (see, e.g. recent sequencing of 236 individuals from 125 distinct human populations by Sudmant et al., 201 a study of 456 geographically diverse high-coverage Y chromosome sequences to infer second strong bottleneck in Y-chromosome lineage by Karmin et al., 202 genotyping and comprehensive analysis of 2,039 samples from rural areas within UK by Leslie et al., 203 as well as many others projects 13,158,167,204–208 ). Parallel improvement of experimental and computational methods will enable studies of ancient populations instead of just a few individuals, and new studies of this kind are emerging now. 11,12,28 The utility of aDNA data will increase with further progress in the genotype-to-phenotype mapping of humans. For the first time, we can anticipate the direct study of evolution for traits that are not associated with the fossil record, such as metabolic and behavioural details. aDNA will provide an important source of information on the origins of cells that harboured DNA thousands of years ago, the age of samples at the time of death, and the environmental influences. Altogether, analysis of aDNA will help us to better understand our world and our role in it.
Funding
I.M. was supported by Swiss Mäxi Foundation grant. H.A. was supported by NIH grants GM098741 and MH091561. E.P. was supported by Russian Science Foundation grant 14-14-01202. P.F. was supported by the Moravian Silesian region projects MSK2013-DT1, MSK2013-DT2, and MSK2014-DT1 and by the Institution Development Program of the University of Ostrava. T.V.T., E.E., and P.P. were supported by NSF Division of Environmental Biology Award 1456634. E.E. was supported by The Royal Society International Exchanges Award (IE140020) and MRC Confidence in Concept Scheme Award 2014-University of Sheffield (Ref: MC_PC_14115). E.R. was supported by Russian Science Foundation grant 14-50-00029.
Acknowledgements
We are grateful to Drs Lana Grinberg, David E. Cobrinik, Roger Jelliffe, and Steven D. Aird for helpful comments. The idea of the article was conceptualized during the Ancient DNA Symposium sponsored by the Okinawa Institute of Science and Technology.
Conflict of interest
None declared.
References
Author notes
Edited by Dr Katsumi Isono

{kind=link}
{kind=link}
{kind=link}
{kind=link}